Sind die Arzneimittel-Zulassungsbehörden gekauft? Ein Beitrag mit Brisanz.
Aus der Naturheilpraxis von René Gräber / Kategorie: Medikamente
Ende Juni 2022 erschien in der „British Medical Journal“ (BMJ) [1] ein besonders interessanter Beitrag, den ich leider erst neulich hatte entdecken können. Es geht um die Finanzierung der Zulassungsbehörden für pharmazeutische Produkte weltweit.
Die Autorin, eine Ärztin und investigative Journalistin, geht der Frage nach, inwieweit FDA, EMA, MHRA etc. von der Pharmaindustrie finanziert werden und welchen Einfluss das auf die Zulassungspraxis hat.
Und der Beitrag beginnt mit einer wenig erfreulichen Aussage, dass nämlich während der letzten Jahrzehnte diese Aufsichtsbehörden mehr und mehr von genau der Industrie finanziert werden, die sie eigentlich beaufsichtigen sollen. Und das heißt dann was?
Geld, Geld und nochmals: Geld
Im Jahr 1992 verabschiedete der US-Kongress den Prescription Drug User Fee Act (PDUFA), der es der Industrie ermöglichte, die US Food and Drug Administration (FDA) direkt durch „Benutzungsgebühren“ zu finanzieren, um die Kosten für die rasche Prüfung von Arzneimittelanträgen zu decken. Mit diesem Gesetz wurde die FDA von einer vollständig aus Steuergeldern finanzierten Einrichtung zu einer Einrichtung, die durch Gelder der Industrie ergänzt wird. Die eingenommenen PDUFA-Nettogebühren sind um das 30-fache gestiegen – von etwa 29 Millionen Dollar im Jahr 1993 auf 884 Millionen Dollar im Jahr 2016.
In Europa wurde die neue EU-weite Zulassungsbehörde, die Europäische Arzneimittel-Agentur (EMA), im Jahr 1995 zu 20 % durch Gebühren der Industrie finanziert. Bis 2010 war dieser Anteil auf 75 % gestiegen; heute sind es 89 %.
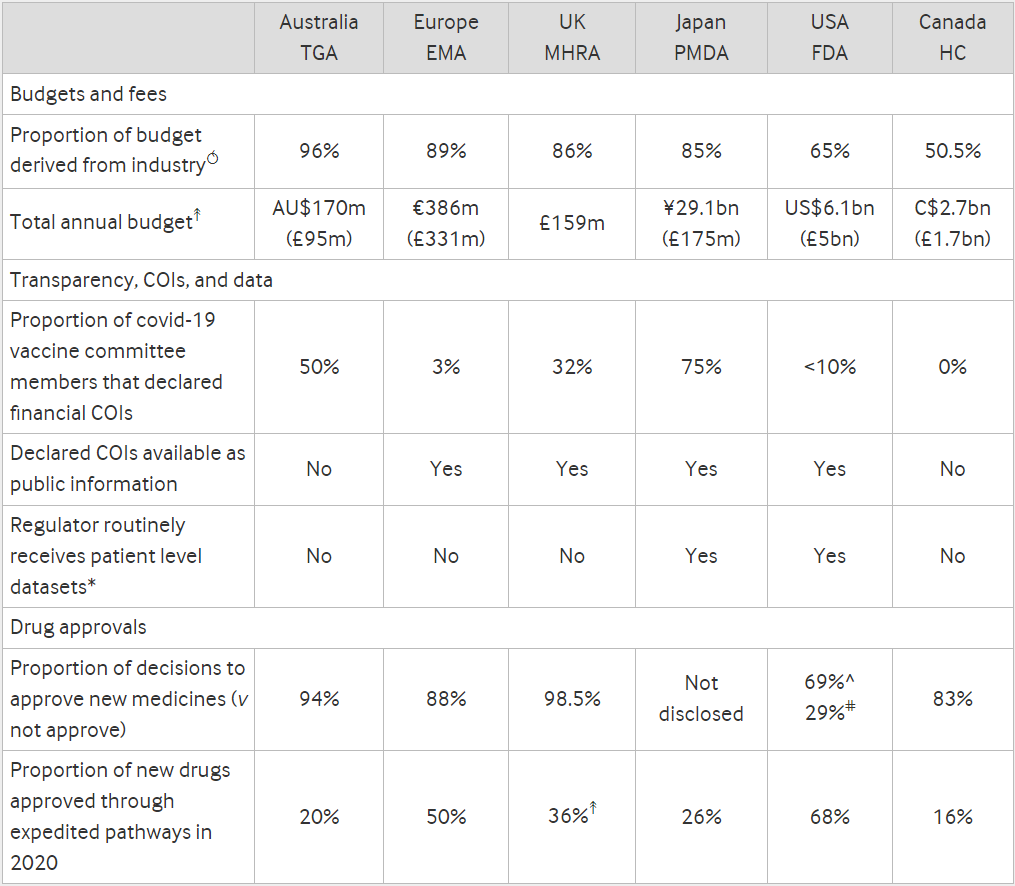
Die Beispiele gehen weiter. Hier eine tabellarische Übersicht aus dem BMJ-Beitrag:
Unschwer zu erkennen ist, dass die australischen Zulassungsbehörden zu 96 % von der Industrie finanziert werden. Oder mit anderen Worten: Die TGA gehört der Pharmaindustrie.
Aber dennoch versteigt sich die TGA zu behaupten, dass diese finanzielle Abhängigkeit keinen Interessenkonflikt darstellt. 96 % Finanzierung und keine Interessenkonflikte? Wenn die „Impfungen“, die auch von der TGA zugelassen wurden, angeblich 95 % Wirksamkeit haben, wieso ist dann eine 96-prozentige Finanzierung „unwirksam“ in Bezug auf Interessenkonflikte? Ist es wirklich wahr, dass profitorientierte Unternehmen, wie die Pharmaindustrie, einfach nur mal Geld aus dem Fenster werfen, ohne davon entsprechende Vorteile zu haben? Wer glaubt wird selig…
Die EMA, MHRA und die japanische Zulassungsbehörde PMDA werden alle zu deutlich über 80 % von der Pharmaindustrie „betrieben“. Die FDA liegt mit 65 % deutlich dahinter, ebenso die kanadischen Zulassungsbehörden mit 50,5 %. Aber auch diese Zahlen sind signifikant und legen Interessenkonflikte nahe.
Wie sehr es Interessenkonflikte gibt und wie sie bereits zutage getreten sind, dass erläutert die Autorin in folgenden Beispielen:
Gekaufte Skandale und institutionelle Korruption
„Doch seit Jahrzehnten stellen Wissenschaftler den Einfluss der Finanzierung auf Zulassungsentscheidungen in Frage, insbesondere nach einer Reihe von Skandalen bei Arzneimitteln und Medizinprodukten – darunter Opioide, Alzheimer-Medikamente, antivirale Grippemittel, Beckennetze, Gelenkprothesen, Brust- und Verhütungsimplantate, Herzstents und Herzschrittmacher. Eine Analyse von drei Jahrzehnten PDUFA in den USA hat gezeigt, wie die Abhängigkeit von den Gebühren der Industrie zu einer Verschlechterung der Beweisstandards beiträgt, was letztlich den Patienten schadet. In Australien haben Experten eine vollständige Überarbeitung der Struktur und Funktion der TGA gefordert und argumentiert, dass die Behörde der Industrie zu nahe gekommen ist.“
Ein Soziologe von der Universität New Jersey nennt dann das Kind beim Namen: Die weitestgehende Finanzierung der Zulassungsbehörden durch die Pharmaindustrie ist nicht nur ein fundamentaler Interessenkonflikt sondern auch ein Paradebeispiel für institutionelle Korruption.
Unter diesen Gesichtspunkten und nach den vorausgegangenen Medizinskandalen war die Zulassung („Notfallzulassung“) der Gen-Injektionen der nächste logische Schritt in dieser Entwicklung.
Und heute sind wir ja bereits so weit, dass neue genbasierte „Impfungen“ überhaupt nicht mehr getestet werden brauchen, höchstens mal an acht Mäusen, weil man ja die Erfahrungen von den „Coronaimpfungen“ eins zu eins auf die neuen Produkte übertragen kann.
Das heißt, die Pharmaindustrie spart Geld und Zeit, gleichgültig wie das Resultat für die Kundschaft aussehen wird. Im Zweifelsfall leugnet man einfach einen Zusammenhang zwischen „Impfung“ und beobachteten Nebenwirkungen. Alles nur Zufall… und zufällig produzieren die „Impfungen“ auch noch Zufälle am laufenden Band. Unter diesen Voraussetzungen kann man das Wort „Nebenwirkungen“ dann auch gleich aus dem Vokabular streichen.
Übrigens: Wenn Sie solche Informationen interessieren, dann fordern Sie unbedingt meinen kostenlosen Praxis-Newsletter „Unabhängig. Natürlich. Klare Kante.“ dazu an:
Transparenz der Daten
Unter diesen Voraussetzungen ist es fast egal, ob die Hersteller oder die Zulassungsbehörden die entsprechenden Zulassungsstudien durchführen. Laut BMJ verlassen sich die meisten Zulassungsbehörden auf die Daten der Hersteller und machen keine eigenen Untersuchungen.
Die TGA sagt zum Beispiel, dass sie ihre Bewertungen des Covid-19-Impfstoffs auf der Grundlage der vom Sponsor des „Impfstoffs“ bereitgestellten Informationen durchführt. In einer FOI-Anfrage vom Mai letzten Jahres erklärte die TGA, dass sie die Quelldaten der Covid-19-Impfstoffversuche nicht gesehen habe.
Wie bitte???
Stattdessen wertete die Behörde die „aggregierten oder gepoolten Daten“ des Herstellers aus. Die TGA verfügt auch nicht über die Datensätze der einzelnen Teilnehmer an den Covid-19-Impfstoffstudien, die sich im Besitz des Impfstoffherstellers befinden.
Oder mit anderen Worten: Die Zulassung – ein kompletter Blindflug. Aber warum großen Aufstand machen, wenn das Ergebnis, die Zulassung der Gen-Injektionen, von Anfang an abgemachte Sache war!?
Von den allen Zulassungsbehörden weltweit erhalten nur die FDA und die PMDA die Patientendaten der einschlägigen Studien. Alle anderen Zulassungsbehörden stochern im Nebel. Aber von der FDA wissen wir ja, dass sie die Patienten- bzw. Studiendaten zum Pfizer-Produkt für 75 Jahre unter Verschluss halten wollte, was aber durch einen Gerichtsbeschluss verhindert wurde.
Ich erwähnte bereits die „Benutzungsgebühren“, die 1992 für die FDA eingeführt wurden. Dieses Geld sollte für zusätzliche Angestellte ausgegeben werden, um die Zulassung für neue Medikamente zu beschleunigen. Denn mehr Angestellte bewältigen mehr Arbeit, so die Logik.
Heute ist das Phänomen der beschleunigten Zulassung bei allen Zulassungsbehörden zu beobachten. Im Jahr 2020 wurden 68 % von neuen Medikamenten in den USA im Schnellverfahren zugelassen, 50 % in Europa und 36 % in Großbritannien. Das Ergebnis dieser Praxis war und ist, dass mehr Medikamente aus Sicherheitsgründen vom Markt genommen werden (müssen), mehr Medikamente einen expliziten Warnhinweis erhalten oder Dosierungsstärken der neuen Substanz aus dem Programm genommen werden müssen.
Aaron Kesselheim, Professor für Medizin am Brigham and Women’s Hospital und an der Harvard Medical School, fügt hinzu, dass beschleunigte Zulassungen im Allgemeinen eine geringere Beweislast für die Wirksamkeit haben.
Kein Wunder also, dass die „Coronaimpfungen“ trotz fehlender Wirksamkeit angeblich eine Wirksamkeit von 95 % haben sollen. Daraus folgt auch, dass es kein Wunder ist, dass niemand mehr von diesen 95 % spricht.
Auf welcher Basis wird dann eine schnelle Zulassung durchgeführt, wenn die Zulassungsbehörde noch nicht einmal die Originaldaten der Studienteilnehmer vom Hersteller erhält?
Die Antwort: Stochern im Nebel und ein blindes Huhn findet auch mal ein Korn. In der Zulassungspraxis sieht das dann so aus:
„Der beschleunigte Zulassungsweg ändert ausdrücklich den zugrundeliegenden ‚Wirksamkeitsstandard‘, indem er die Zulassung auf der Grundlage von Änderungen eines Surrogatmaßes erlaubt, das nicht gut validiert ist und den klinischen Nutzen nur mit einiger Wahrscheinlichkeit vorhersagen kann“, sagt Kesselheim, der 2021 aus Protest gegen die Zulassung eines umstrittenen Alzheimer-Medikaments durch die Behörde aus einem FDA-Beratungsausschuss ausgetreten ist. Nachdem der Ausschuss gegen die Zulassung gestimmt hatte, änderte die FDA die Zielrichtung und genehmigte Aducanumab im Rahmen eines beschleunigten Zulassungsverfahrens auf der Grundlage des umstrittenen Surrogatmaßes der Senkung der sichtbaren ?-Amyloid-Proteinspiegel.
Oder mit anderen Worten: Surrogate oder Stellvertreterwerte sind der Trick, mit dem man alles und jedes zulassen kann, wenn der „Zulassungsbeschleuniger“, die „Benutzungsgebühr“, ausreichend hoch ausfällt.
Da soll noch jemand sagen, Pharmaindustrie und Behörden seien nicht „innovativ“.
Die Sache mit dem Drehtüreffekt
Es ist seit langem bekannt, das „verdiente“ Funktionäre in den Zulassungsbehörden lukrative Angebote von der Pharmaindustrie bekommen. Bei der FDA zum Beispiel waren zwischen 2006 und 2019 neun von zehn ehemaligen FDA-Chefs in der Lage, gut dotierte Posten in der Pharmaindustrie zu ergattern. Der elfte ehemalige FDA-Leiter von 2019-2021, Stephen Hahn, wurde Chief Medical Officer bei einer Risikokapitalgesellschaft, die Moderna ins Leben gerufen hatte.
Natürlich alles nur Zufall…
Der momentane FDA-Leiter, Robert Califf, scheint auch kein unbeschriebenes Blatt zu sein. Ihm werden ebenfalls enge Verbindungen zur pharmazeutischen Industrie nachgesagt. So erhielt er angeblich 2,7 Millionen Dollar von Verily Life Sciences. Und 2021 war er Mitglied des Vorstands von zwei pharmazeutischen Firmen (AmyriAD und Centessa Pharmaceuticals).
Bestimmt nur Zufall…
Die Liste der Beispiele lässt sich beliebig verlängern. Inzwischen dürfte es schwer fallen, jemanden zu finden, der in dieser Position keine Verflechtungen zur Industrie hat. Diese Verfilzung scheint heute die Norm („new normal“?) zu sein. Nur hoffnungslose Optimisten würden auch hier alles nur als „Zufall“ interpretieren.
Fazit
Der zuvor zitierte Soziologe von der Universität von New Jersey, Donald Light, fast die Situation treffend zusammen:
„Das ist das Gegenteil einer vertrauenswürdigen Organisation, die unabhängig und streng die Arzneimittel bewertet. Sie sind nicht rigoros, sie sind nicht unabhängig, sie sind selektiv und sie halten Daten zurück. Ärzte und Patienten müssen begreifen, dass man den Arzneimittelbehörden nicht trauen kann, solange sie von der Industrie finanziert werden.“
Übrigens: Wenn Sie solche Informationen interessieren, dann fordern Sie unbedingt meinen kostenlosen Praxis-Newsletter „Unabhängig. Natürlich. Klare Kante.“ dazu an:
Ihre Hilfe für die Naturheilkunde und eine menschliche Medizin! Dieser Blog ist vollkommen unabhängig, überparteilich und kostenfrei (keine Paywall). Ich (René Gräber) investiere allerdings viel Zeit, Geld und Arbeit, um ihnen Beiträge jenseits des "Medizin-Mainstreams" anbieten zu können. Ich freue mich daher über jede Unterstützung!
Helfen Sie bitte mit! Setzen Sie zum Beispiel einen Link zu diesem Beitrag oder unterstützen Sie diese Arbeit mit Geld. Für mehr Informationen klicken Sie bitte HIER.
https://naturheilt.com/blog/wp-content/uploads/2023/09/medikamente_tabletten_pixabay.com-Matvevna.jpg8531280René Gräberhttps://naturheilt.com/blog/wp-content/uploads/2025/07/logonatur.pngRené Gräber2023-09-05 16:47:062023-09-22 11:26:10Sind die Arzneimittel-Zulassungsbehörden gekauft? Ein Beitrag mit Brisanz.
Um unsere Webseite für Sie optimal zu gestalten und fortlaufend verbessern zu können, verwenden wir Cookies. Durch die weitere Nutzung der Webseite stimmen Sie der Verwendung von Cookies zu
Wir können Cookies anfordern, die auf Ihrem Gerät eingestellt werden. Wir verwenden Cookies, um uns mitzuteilen, wenn Sie unsere Websites besuchen, wie Sie mit uns interagieren, Ihre Nutzererfahrung verbessern und Ihre Beziehung zu unserer Website anpassen.
Klicken Sie auf die verschiedenen Kategorienüberschriften, um mehr zu erfahren. Sie können auch einige Ihrer Einstellungen ändern. Beachten Sie, dass das Blockieren einiger Arten von Cookies Auswirkungen auf Ihre Erfahrung auf unseren Websites und auf die Dienste haben kann, die wir anbieten können.
Notwendige Website Cookies
Diese Cookies sind unbedingt erforderlich, um Ihnen die auf unserer Webseite verfügbaren Dienste und Funktionen zur Verfügung zu stellen.
Da diese Cookies für die auf unserer Webseite verfügbaren Dienste und Funktionen unbedingt erforderlich sind, hat die Ablehnung Auswirkungen auf die Funktionsweise unserer Webseite. Sie können Cookies jederzeit blockieren oder löschen, indem Sie Ihre Browsereinstellungen ändern und das Blockieren aller Cookies auf dieser Webseite erzwingen. Sie werden jedoch immer aufgefordert, Cookies zu akzeptieren / abzulehnen, wenn Sie unsere Website erneut besuchen.
Wir respektieren es voll und ganz, wenn Sie Cookies ablehnen möchten. Um zu vermeiden, dass Sie immer wieder nach Cookies gefragt werden, erlauben Sie uns bitte, einen Cookie für Ihre Einstellungen zu speichern. Sie können sich jederzeit abmelden oder andere Cookies zulassen, um unsere Dienste vollumfänglich nutzen zu können. Wenn Sie Cookies ablehnen, werden alle gesetzten Cookies auf unserer Domain entfernt.
Wir stellen Ihnen eine Liste der von Ihrem Computer auf unserer Domain gespeicherten Cookies zur Verfügung. Aus Sicherheitsgründen können wie Ihnen keine Cookies anzeigen, die von anderen Domains gespeichert werden. Diese können Sie in den Sicherheitseinstellungen Ihres Browsers einsehen.
Andere externe Dienste
Wir nutzen auch verschiedene externe Dienste wie Google Webfonts, Google Maps und externe Videoanbieter. Da diese Anbieter möglicherweise personenbezogene Daten von Ihnen speichern, können Sie diese hier deaktivieren. Bitte beachten Sie, dass eine Deaktivierung dieser Cookies die Funktionalität und das Aussehen unserer Webseite erheblich beeinträchtigen kann. Die Änderungen werden nach einem Neuladen der Seite wirksam.











Hinterlasse einen Kommentar
An der Diskussion beteiligen?Hinterlasse uns deinen Kommentar!